Labs and Groups
Kothary Lab
Kothary Lab
Team Leader

Rashmi Kothary
Senior Scientist, Regenerative MedicineWhat We Do
Our laboratory is interested in understanding the mechanisms which maintain/alter muscle and nervous system integrity. In parallel, we are assessing ways to alleviate the pathology of neuromuscular diseases.
Research Activities
Our research program is focused on understanding the mechanisms which maintain or alter muscle and nervous system integrity and assessing ways to alleviate the pathology of neuromuscular disease, specifically Spinal Muscular Atrophy (SMA) and Multiple Sclerosis (MS). Our research builds on a multidisciplinary approach that includes mouse modeling, genetics, cell biology, and biochemistry.
We are studying the pathogenesis of the genetic disease SMA, which is characterized by progressive degeneration of the large motor neurons in the spinal cord and consequent muscular atrophy. Accumulating evidence, much of it from the Kothary lab, shows that abnormalities in other cell types and organs also contribute to the overall disease burden in SMA. We have generated mouse models of SMA with severe/intermediate phenotypes. These mice are now used in many labs throughout the world. Work from our lab led directly to the use of ROCK inhibitors (which modulate cytoskeletal actin filaments) as a therapeutic for SMA. We have also shown glucose metabolism and pancreatic developmental defects in SMA. This work points to the need for metabolic assessment and therapeutic intervention when considering clinical care management for SMA patients. We have demonstrated intrinsic muscle defects in SMA and most recently described immune organ defects. This latter work raises the question of whether immune dysfunction and neuroinflammation contribute to disease pathogenesis. In other research, we discovered liver abnormalities and lipid metabolic defects in SMA. As therapies for SMA come on the market and children live longer, the expectation is that the extended life span will unmask additional health problems and co-morbidities that will need attention. Collectively, our work highlights SMA as a multi-organ disorder. These unanticipated discoveries of SMA pathogenesis were initially deemed controversial yet are now widely accepted as critical drivers of overall SMA pathology, a discovery initiated and championed by the Kothary laboratory.
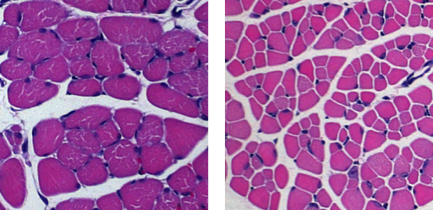
Skeletal muscle atrophy in SMA mice.

The other major area of interest is in MS, where immune cells of the body attack myelin, the protective covering of neurons. Loss of myelin results in defects in electrical conductance in the neurons and subsequent problems including difficulties in maintaining balance and proper gait. Oligodendrocytes are responsible for repairing the damaged myelin. Unfortunately, in many progressive forms of MS, repair is attenuated due to the presence of inhibitory factors at the site of damage. We have developed methods to study oligodendrocyte differentiation in culture that are now widely used by the field. We are identifying key inhibitory factors at MS lesions that prevent the full maturation of oligodendrocytes and halt remyelination in damaged areas of the CNS. We are investigating the molecular mechanisms underpinning the failure to repair damage in MS. We have shown that specific microRNAs are elevated at MS lesions and inhibit the maturation of oligodendrocytes. We are testing whether acute antagonism of these microRNAs have therapeutic benefit in a mouse model of MS. The findings will have an impact on development of future therapeutic strategies targeting repair of damaged myelin.
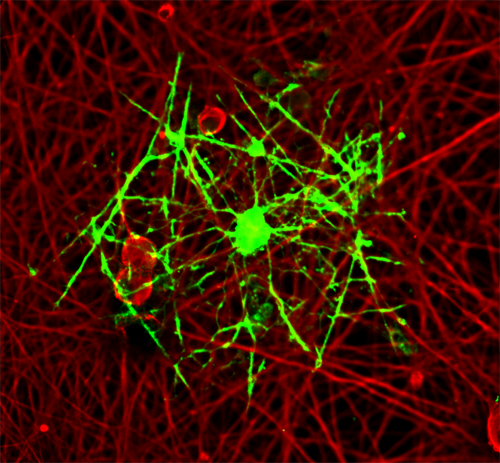
An oligodendrocyte myelinating dorsal root gangion neurite in a co-culture system.
We are studying the pathogenesis of the genetic disease SMA, which is characterized by progressive degeneration of the large motor neurons in the spinal cord and consequent muscular atrophy. Accumulating evidence, much of it from the Kothary lab, shows that abnormalities in other cell types and organs also contribute to the overall disease burden in SMA. We have generated mouse models of SMA with severe/intermediate phenotypes. These mice are now used in many labs throughout the world. Work from our lab led directly to the use of ROCK inhibitors (which modulate cytoskeletal actin filaments) as a therapeutic for SMA. We have also shown glucose metabolism and pancreatic developmental defects in SMA. This work points to the need for metabolic assessment and therapeutic intervention when considering clinical care management for SMA patients. We have demonstrated intrinsic muscle defects in SMA and most recently described immune organ defects. This latter work raises the question of whether immune dysfunction and neuroinflammation contribute to disease pathogenesis. In other research, we discovered liver abnormalities and lipid metabolic defects in SMA. As therapies for SMA come on the market and children live longer, the expectation is that the extended life span will unmask additional health problems and co-morbidities that will need attention. Collectively, our work highlights SMA as a multi-organ disorder. These unanticipated discoveries of SMA pathogenesis were initially deemed controversial yet are now widely accepted as critical drivers of overall SMA pathology, a discovery initiated and championed by the Kothary laboratory.
Skeletal muscle atrophy in SMA mice.
The other major area of interest is in MS, where immune cells of the body attack myelin, the protective covering of neurons. Loss of myelin results in defects in electrical conductance in the neurons and subsequent problems including difficulties in maintaining balance and proper gait. Oligodendrocytes are responsible for repairing the damaged myelin. Unfortunately, in many progressive forms of MS, repair is attenuated due to the presence of inhibitory factors at the site of damage. We have developed methods to study oligodendrocyte differentiation in culture that are now widely used by the field. We are identifying key inhibitory factors at MS lesions that prevent the full maturation of oligodendrocytes and halt remyelination in damaged areas of the CNS. We are investigating the molecular mechanisms underpinning the failure to repair damage in MS. We have shown that specific microRNAs are elevated at MS lesions and inhibit the maturation of oligodendrocytes. We are testing whether acute antagonism of these microRNAs have therapeutic benefit in a mouse model of MS. The findings will have an impact on development of future therapeutic strategies targeting repair of damaged myelin.
An oligodendrocyte myelinating dorsal root gangion neurite in a co-culture system.
Selected Publications
Base editing as a genetic treatment for spinal muscular atrophy. Alves CRR, Ha LL, Yaworski R, Lazzarotto CR, Christie KA, Reilly A, Beauvais A, Doll RM, de la Cruz D, Maguire CA, Swoboda KJ, Tsai SQ, Kothary R, Kleinstiver BP. bioRxiv. 2023 Jan 21:2023.01.20.524978. doi: 10.1101/2023.01.20.524978. Preprint. PMID: 36711797 Free PMC article.
Central and peripheral delivered AAV9-SMN are both efficient but target different pathomechanisms in a mouse model of spinal muscular atrophy. Reilly A, Deguise MO, Beauvais A, Yaworski R, Thebault S, Tessier DR, Tabard-Cossa V, Hensel N, Schneider BL, Kothary R. Gene Ther. 2022 Sep;29(9):544-554. doi: 10.1038/s41434-022-00338-1. Epub 2022 Apr 25. PMID: 35462564
SMN Depleted Mice Offer a Robust and Rapid Onset Model of Nonalcoholic Fatty Liver Disease. Deguise MO, Pileggi C, De Repentigny Y, Beauvais A, Tierney A, Chehade L, Michaud J, Llavero-Hurtado M, Lamont D, Atrih A, Wishart TM, Gillingwater TH, Schneider BL, Harper ME, Parson SH, Kothary R. Cell Mol Gastroenterol Hepatol. 2021;12(1):354-377.e3. doi: 10.1016/j.jcmgh.2021.01.019. Epub 2021 Feb 2. PMID: 33545428 Free PMC article.
MiRNA-145-5p prevents differentiation of oligodendrocyte progenitor cells by regulating expression of myelin gene regulatory factor. Kornfeld SF, Cummings SE, Fathi S, Bonin SR, Kothary R. J Cell Physiol. 2021 Feb;236(2):997-1012. doi: 10.1002/jcp.29910. Epub 2020 Jun 30. PMID: 32602617
Inhibitory milieu at the multiple sclerosis lesion site and the challenges for remyelination. Galloway DA, Gowing E, Setayeshgar S, Kothary R. Glia. 2020 May;68(5):859-877. doi: 10.1002/glia.23711. Epub 2019 Aug 23. PMID: 31441132 Review.
Central and peripheral delivered AAV9-SMN are both efficient but target different pathomechanisms in a mouse model of spinal muscular atrophy. Reilly A, Deguise MO, Beauvais A, Yaworski R, Thebault S, Tessier DR, Tabard-Cossa V, Hensel N, Schneider BL, Kothary R. Gene Ther. 2022 Sep;29(9):544-554. doi: 10.1038/s41434-022-00338-1. Epub 2022 Apr 25. PMID: 35462564
SMN Depleted Mice Offer a Robust and Rapid Onset Model of Nonalcoholic Fatty Liver Disease. Deguise MO, Pileggi C, De Repentigny Y, Beauvais A, Tierney A, Chehade L, Michaud J, Llavero-Hurtado M, Lamont D, Atrih A, Wishart TM, Gillingwater TH, Schneider BL, Harper ME, Parson SH, Kothary R. Cell Mol Gastroenterol Hepatol. 2021;12(1):354-377.e3. doi: 10.1016/j.jcmgh.2021.01.019. Epub 2021 Feb 2. PMID: 33545428 Free PMC article.
MiRNA-145-5p prevents differentiation of oligodendrocyte progenitor cells by regulating expression of myelin gene regulatory factor. Kornfeld SF, Cummings SE, Fathi S, Bonin SR, Kothary R. J Cell Physiol. 2021 Feb;236(2):997-1012. doi: 10.1002/jcp.29910. Epub 2020 Jun 30. PMID: 32602617
Inhibitory milieu at the multiple sclerosis lesion site and the challenges for remyelination. Galloway DA, Gowing E, Setayeshgar S, Kothary R. Glia. 2020 May;68(5):859-877. doi: 10.1002/glia.23711. Epub 2019 Aug 23. PMID: 31441132 Review.